
The 5 drug development phases
Category | CDMO Services
Drug development is undeniably complex.
From molecule discovery to FDA review and safety monitoring to commercialization, there are many opportunities for things to go right and for things to go wrong. In 2022, the FDA approved 37 new drugs, or “novel” drugs .1 But that doesn’t account for the many others that didn’t make the cut. In fact, the overall probability of success for new molecular entities is only 12%.2
To be deemed a “success,” a new drug must make it through five specific phases: 1) discovery and development, 2) preclinical research, 3) clinical research, 4) FDA review, and 5) safety monitoring.
Below, we explore each step in more detail.

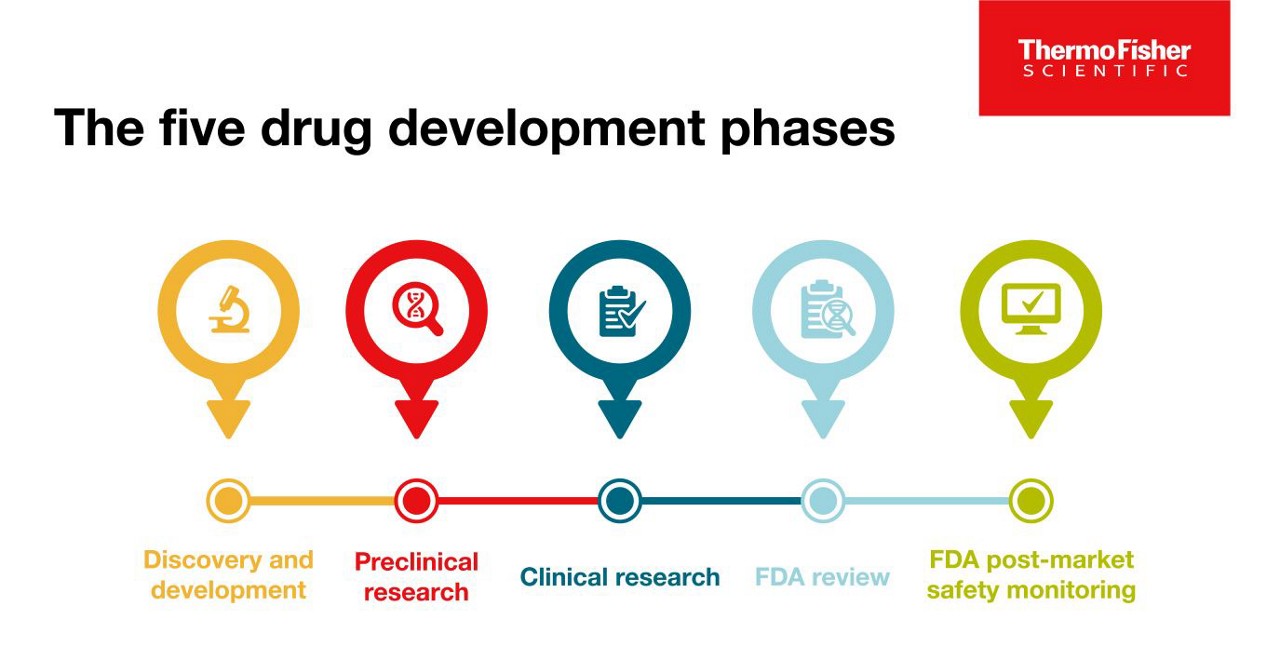
Phase One: Discovery and development
Long before any drug development and manufacturing work can be done, researchers must identify or “discover” a specific molecule — typically a DNA sequence, RNA molecule, protein, or metabolite — that plays a crucial role in a disease state and can be targeted by a drug to produce beneficial and therapeutic effects. After that discovery, researchers can then search for a compound or compounds that interact with the target molecule and have the potential to become drug candidates. A variety of compounds are usually identified as potential candidates, so researchers must conduct a series of experiments on each one to gauge their performance and viability as the final, most effective drug substance. Typically, they assess factors such as absorption, administration, side effects, and potential interactions. After those experiments are finalized, and the most promising compounds are isolated, the preclinical research phase can begin.
“The purpose of preclinical research is largely to assess whether a compound has the potential to cause serious harm.”
Phase Two: Preclinical research
Before any compound can be tested in humans, researchers must conduct preclinical research either in vitro (i.e., in a test tube) or in vivo (i.e., in an animal). The purpose of preclinical research is largely to assess whether a compound has the potential to cause serious harm. Additionally, candidates undergo testing for pharmacodynamics and pharmacokinetics, examining what the drug does to the body and what the body does to the drug. Preclinical research also plays a crucial role in determining pharmaceutical formulation development, including such factors as stability, bioavailability, and administration method. All preclinical research must comply with the FDA’s good laboratory practice (GLP) regulations, which set the standard for data quality, integrity, and reliability. After a drug successfully navigates through preclinical research, it’s ready to be tested in humans.
Phase Three: Clinical research
Clinical research via clinical trials is the next step in drug development, and it serves to test the safety and efficacy of compounds in humans. Clinical research is typically divided into four phases: phase I, phase II, phase III, and phase IV. According to the FDA, phase I involves 20 to 100 healthy volunteers or individuals with the disease or condition, phase II involves up to several hundred people with the disease or condition, phase III involves 300 to 3,000 people with the disease or condition, and phase IV involves several thousand people with the disease or condition. If a drug compound successfully demonstrates safety and efficacy during clinical trials, it can advance to FDA review. As previously stated, it’s important to note that only a small percentage of new drugs that enter clinical trials eventually receive FDA approval. Factors such as lack of efficacy and safety issues often contribute to this low success rate.
“If the drug is deemed safe and effective for its intended use, the FDA grants approval to manufacture, market, and distribute the drug in the United States.”
Phase Four: FDA review
Once a drug moves through phase I, phase II, phase III, and phase IV clinical trials, it advances to FDA review, where a team of experts including doctors, chemists, statisticians, microbiologists, and pharmacologists review a drug compound’s safety and efficacy findings from its clinical trials. When a biotechnology or pharmaceutical company seeks FDA review, they must submit a New Drug Application (NDA) for drugs or a Biologics License Application (BLA) for biologics. Then, the FDA must accept the application and assign a team of experts to evaluate its case. Together, the team reviews the clinical research — including patient outcomes, potential adverse effects, and the drug’s risk-benefit analysis. If the drug is deemed safe and effective for its intended use, the FDA grants approval to manufacture, market, and distribute the drug in the United States.
Phase Five: FDA post-market safety monitoring
Although clinical research serves to evaluate a drug’s safety and efficacy in a relatively small pool of volunteers, it’s possible that new concerns may arise in the general population after its approval. That’s where FDA post-market safety monitoring, or post-market surveillance, comes into play. The FDA has several programs in place to assist with post-market safety monitoring, including MedWatch and MedSun. MedWatch allows healthcare professionals and consumers to report serious problems with medical products, whereas MedSun works collaboratively with the clinical community to identify, understand, and solve problems specifically related to the use of medical devices. Additionally, the FDA conducts routine inspections of a drug product’s manufacturing facilities to ensure they’re compliant with regulatory standards, and they also monitor drug advertisements and labeling to ensure biotechnology or pharmaceutical companies don’t make any false or misleading claims.
A CDMO partner can help companies navigate through drug development
On average, it takes 10-15 years and $2.6 billion to develop one new medicine, including the cost of many failures.3 But biotechnology and pharmaceutical companies don’t have to navigate the drug development journey alone. They can partner with a CDMO, or contract development and manufacturing organization, to access advanced capabilities and resources as well as industry expertise in drug development, manufacturing, and regulatory compliance. Outsourcing key functions such as discovery and development, preclinical and clinical research, and safety monitoring to a CDMO enables biotech and pharmaceutical companies to concentrate on their core strengths and priorities while ensuring compliance with regulatory requirements.
Contact us to learn more about what makes a quality CDMO, or download our whitepaper below.
View references

More on the topic

Blog post
CROs vs CMOs, and CDMOs: What’s the difference between the three?
CROs, CMOs, and CDMOs all help biotechnology and pharmaceutical companies with drug development and manufacturing, but what’s the difference between the three?
Read Blog
Blog post
What is a CDMO?
Learn how CDMOs (contract development and manufacturing organizations) work with pharma companies, and the top considerations companies should have when choosing a CDMO partner.