
Navigating Cell & Gene Therapy Regulations: How Does Your CDMO Match Up?
December 17, 2020 (7 minute read)

Category | Advanced therapy

Cell and gene therapy (CGT) developers today face an added challenge in their quest to bring a product through clinical trials and to the market: disparate classifications, guidelines, and requirements from regulatory bodies in the United States and the European Union. As regulators gain experience with the growing variety of CGT products and processes, however, significant differences in regulatory guidance are likely to ease. Both regions already offer similar opportunities for interaction throughout the development spectrum and fast-tracked review and approval. Other areas likely to undergo regulatory harmonization in the world of CGTs include requirements for source materials, guidelines for lab- and clinic-based validations, and guidance for at-scale product manufacturing.
TIP: In the US, key laws governing the development of biologics and CGTs include Section 505 of the Food, Drug, and Cosmetic Act (1938) and Sections 351 and 361 of the Public Health Service Act (1944).
1. Expect converging guidance regarding raw source material acquisition and testing. Currently, the European Medicines Agency (EMA) and the US Food and Drug Administration (FDA) have different definitions of raw materials and components for CGTs and other biologic products, resulting in distinct expectations for documentation of a substance’s origin and specifications. Moving forward:
In the meantime: A risk–benefit assessment per ICH Q9 on the risk of the starting and/or raw material to the product and process should be considered to mitigate uncertainties and ensure a safe and quality product.
TIP: In the EU, key legislation governing the development of advanced therapy medicinal products (ATMPs) includes ATMP regulation 1394/2007/EC and Directive 2009/120/EC.
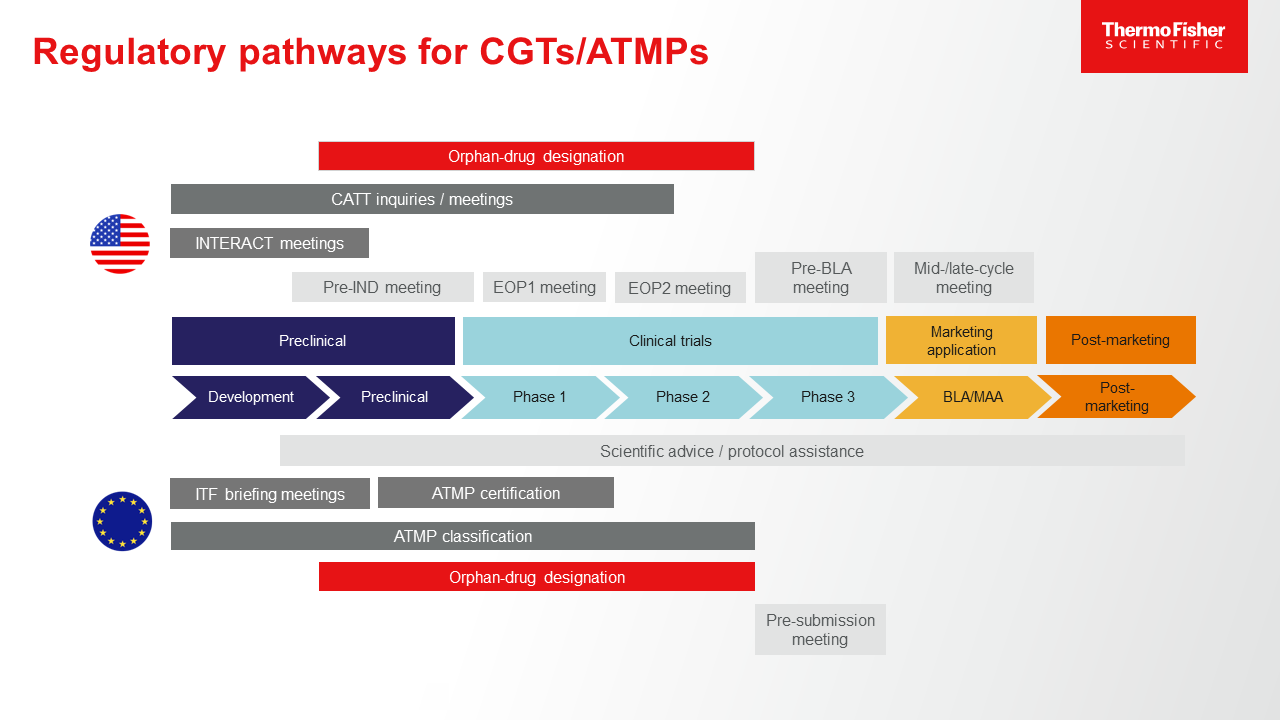
2. Prepare for tightened expectations on preclinical and clinical validation and safety documentation.The challenge presented by CGTs (known as advanced therapy medicinal products or ATMPs in EU legislation and guidance) in the world of preclinical and clinical research is that, for some novel therapies, the best way to demonstrate safety, purity, potency, on-target effect, lack of toxicity, pharmacokinetic/pharmacodynamic response, and other factors is sometimes unclear. This makes identification of the right assays, outcome measures, endpoints, and safety monitoring tests a nebulous process—one that, at least at present, is often discussed and selected on a case-by-case basis using whatever knowledge is available. As drug delivery methods, mechanisms of action, and CGT source materials become more common and familiar to regulatory experts, expect:
In the meantime: Early and routine consultation with both the EMA and FDA can help address questions related to classification, analytics, and clinical and manufacturing concerns.
3. Anticipate clearer expectations for manufacturing parameters. Despite differences in classification and source material requirements, overall expectations for manufacturing parameters are already fairly similar in the EU and the US. It is reasonable to expect:
In the meantime: CDMOs that have experience producing approved CGTs are especially valuable in overcoming manufacturing challenges and easing the approval process for novel therapies, by relying on prior work to help inform and quickly advance new processes.
In conclusion, more formal and convergent guidance on CGT development and production stands to benefit both biopharmaceutical companies and patients, but it will require ongoing data acquisition and the evolution of regulatory pathways, which take time. As regulatory bodies gain experience in the coming years with a wide variety of CGTs and their respective attributes, developers will likely receive increasingly similar, standardized guidance intended to clarify research efforts and streamline the development and application process across regions. Despite the persistence of some clear jurisdictional differences, as seen in the world of nonbiologic drug development, more consistent expectations are likely to trickle into other regions’ regulatory guidance for CGTs as well. In the meantime, understanding the spirit of US and EU regulations—which hinge on protecting patient safety—will be a critical guidepost for CGT sponsors.
CLICK HERE to view our recent webinar, “Flexible regulatory pathways and key CMC considerations to commercialize cell and gene therapy products,” with industry experts Monica Commerford and Clara Ferloni.